Advanced Search
Introduction
Customise your own search query and retrieve a set of ENA records tailored to your search criteria.
All searches are performed against a subset of the archive specified by the Data type you choose to search against. You can then build your search query to specify what data you are looking for and select what fields you want to retrieve from your search. There are additional options to include/exclude specific datasets as well as filter the number of results you wish to return.
If you want to access the same data programmatically, you can copy the produced curl request and run this yourself against the ENA Portal API.
Data Types
Each data type is a subset of the data and metadata held within the ENA.
You must specify which datatype you wish to search across before you can narrow down your search results with any additional criteria.
What is in each data type?
Data Type |
Description |
API Result type |
Studies |
All studies in ENA. Studies can be
searched by the study metadata, e.g.
title or by related taxonomy of data.
|
study |
Studies used
for raw reads
|
All studies that hold raw read datasets.
Searching across this data type can give
you information on these studies and can
be filtered down by sample metadata
(information about the collected samples
in the study).
|
read_study |
Studies used
for nucleotide
sequence
analyses from
reads
|
All studies that hold any nucleotide
analyses. Searching across this data type
can give you information on these studies
and can be filtered down by sample
metadata (information about the collected
samples in the study).
|
analysis_study |
Samples |
All samples in ENA. Samples can be
searched by sample metadata.
|
sample |
Experiments
used for raw
reads
|
All metadata associated with sequencing
event used to generate raw reads
submitted to ENA.
|
read_experiment |
Raw reads |
All raw read datasets in ENA. This
datatype can be filtered by sample
metadata (information about the collected
sample) as well as return directly
downloadable files.
|
read_run |
Nucleotide
sequence
analyses from
reads
|
All nucleotide analyses held in ENA.
This datatype can be filtered by sample
metadata (information about the collected
sample) as well as return directly
downloadable files.
|
analysis |
Genome
assemblies
|
All genome assembly records (excluding
primary or binned metagenomes which are
considered analyses). This datatype can
only be filtered by assembly metadata
e.g. project accession or taxonomy.
|
assembly |
Genome
assembly
contig sets
(WGS)
|
All genome assembly contig sets in ENA.
This datatype can be filtered by sample
metadata (information about the collected
sample).
|
wgs_set |
Transcriptome
assembly
contig sets
(TSA)
|
All transcriptome assembly contig sets in
ENA. This datatype can be filtered by
sample metadata (information about the
collected sample).
|
tsa_set |
Nucleotide
sequences
|
All assembled and annotated sequences
publicly available in ENA (including high
level sequences from assembly submissions
e.g. fully assembled chromosomes).
|
sequence |
Protein coding
sequences
|
All protein coding sequences publicly
available in ENA
|
coding |
Non-coding
sequences
|
All non coding sequences publicly
available in ENA
|
noncoding |
Taxonomic
classification
|
All taxa and their tax IDs. Search for a
specific taxa or look for all taxa below
a tax node.
|
taxon |
Search Query
To build a query you need to create a list of rules that the resulting data sets should be restricted to.
This is done by clicking any relevant metadata types you would like to restrict (listed as buttons on the left) then selecting the relevant filters and specifying the desired restrictions for those:
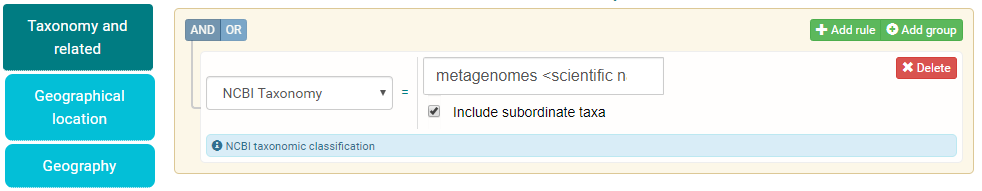
When specifying taxonomy in your search, you can include all subordinate taxa within that tax tree and when searching by geographical range you can interactively drag the box or circle over the desired region to automatically fill out the location range.
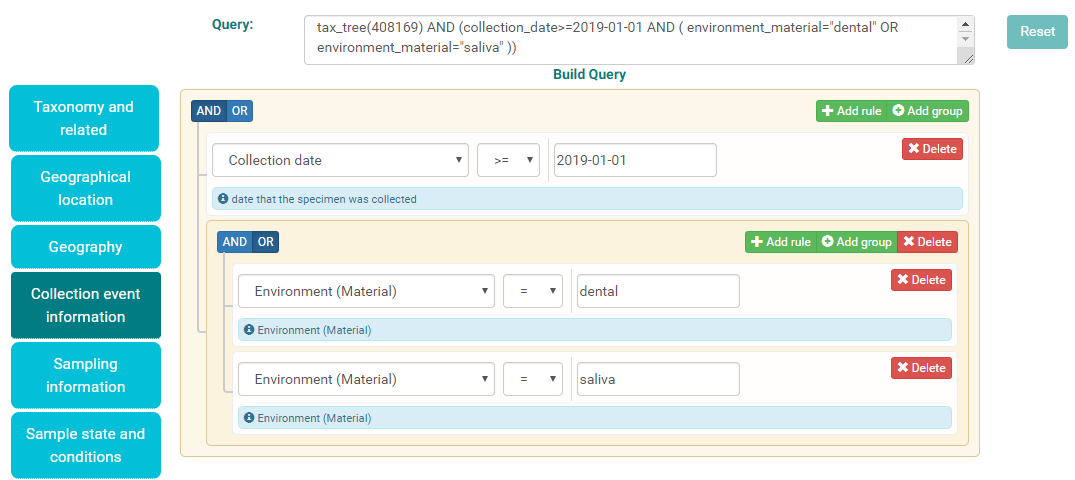
These rules can be grouped and nested within AND or OR logical statements. For example, a query for all metagenomic analyses where the sample was collected after 01 Jan 2019 AND the environmental material is either dental OR saliva would look as follows:
Inclusion/Exclusion of datasets
If there are any known public datasets that do or do not fit the criteria you have specified that you wish to include or exclude from the results, you can list the accessions in a comma separated list here (with no spaces).
Third party annotation
Third party curations are additional metadata associated to INSDC records, which have been submitted by the user community to the ELIXIR Data Clearinghouse. The original records have not been altered.
Return Fields
By default, you will receive the accession and description/title of the main datatype you are searching against. If you wish to customise the metadata which your search will return, you can manually select your search return fields from a list of all indexed fields for the specified datatype.
Select and order fields
To select fields you would like returned from your search, drag across any desired fields from the Available Fields list to the Selected Fields list. Alternatively, use the arrow buttons in the middle to move fields across from one list to the other.
The order of the Selected Fields list will define the order that you receive those metadata from your search. To specify the return order of these fields, you can drag and drop these into the desired order.
Field sets
Field sets are a pre-defined set of fields that can be returned together and are available for some data types. For example, for the analysis datatype, you can toggle the ‘Submitted Files’ field set which can be used to return all relevant fields relating to the original set of submitted files (e.g. this set includes the aspera, ftp and galaxy links for the submitted files, the size of the files (in bytes) and the files’ md5 checksums).
Data Filters
Offset
You can specify an offset for the number of records you would like to skip from the beginning of your search. This can be used to view results beyond the maximum number of records that can be viewed in the results table (100 000) or to break up queries that result in a large number of records into smaller batches.
If you do not wish to skip any records, you can leave this field blank or enter an offset of ‘0’.
Limit
You can specify a data limit for the maximum number of records you would like to retrieve from your search.
If you wish to fetch the full result set enter ‘0’. Leaving the limit field blank applies the default limit of 100 000. For large result sets, to get all records please download the report (JSON/TSV) or copy and run the curl command outside of the browser.
Download ENA records
Here you can download the ENA records resulting from your search.
This will download the whole ENA record stored for each of the results. If you wish to only download the fields returned that were specified in your search, use one of the Download report options (JSON or TSV).
XML records
XML records are available for all standard metadata objects held within ENA (all results with the exception of sequence records).
XML records hold all the metadata for each object concatenated into a single bulk XML file. These XML metadata records are formatted in the standard ENA XML format (the same XML format that is used for data submission and for data to be displayed in the browser).
FASTA records
FASTA records hold all sequences resulting from your search concatenated into one FASTA file. FASTA records are only available when searching against sequence datatypes.
EMBL records
EMBL flatfile records hold all sequences resulting from your search and their annotation (if available) concatenated into a single EMBL flat file. EMBL records are only available when searching against sequence datatypes.
Download results report
This feature allows you to download all the results from your search in the format of a JSON or TSV file. Any data filters set by you will apply here.
Download associated data files
Pre-Conditions
To see file download columns in your results, you have to search against either the analysis or read_run data types and select the relevant fields that end with ‘_ftp’.
For example:
Data Type = analysis and fields = submitted_ftp
Data Type = read_run and fields = fastq_ftp / sra_ftp / submitted_ftp
Download data files
You can download the data files resulting from your search in one of four ways:
The ENA File Downloader is a new Java based command line application. You have to submit one or more comma separated accessions, or a file with accessions that you want to download data for. This tool allows downloading of read and analysis files, using FTP or Aspera. It has an easy to use interactive interface and can also create a script which can be run programmatically or integrated with pipelines.
Download the latest version from ENA Tools.
or from github at ena-ftp-downloader.
You can download a single file by clicking on its link in the FASTQ FTP, SRA FTP, or SUBMITTED FTP column.
You can select one or more files using the check boxes, and either download a downloader script which you can run separately or as individual files using the “Get download script” or “Download selected files” links above the table.
You can download a script that contains the download requests for ALL files resulting from your search by clicking the download icon in the column header.

Tips:
If you wish to exclude any records from your search results before you download all the resulting files, you can go back and list these in the “Exclude Accessions” field and then repeat the search.
If you selected multiple files and clicked the “Individually” link but only the first file is downloading, this could be because your browser is restricting multiple download pop-ups. Look for a browser warning or confirmation dialog to allow this.
If selecting many files and using the download “Individually” option, you may wish to change the default download location of your browser. Look in your browser settings for this.
You can also download files using a terminal from ENA using enaBrowserTools.